
Metabolically engineered bacteria as light-controlled living therapeutics for anti-angiogenesis tumor therapy†
Guping
Tang  *c
and
Bin
Liu
*ad
*c
and
Bin
Liu
*ad
*c
and
Bin
Liu
*ad
Abstract
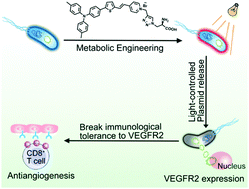
A living therapeutic system based on attenuated Salmonella was developed via metabolic engineering using an aggregation-induced emission (AIE) photosensitizer MA. The engineered bacteria could localize in the tumor tissues and continue to colonize and express exogenous genes. Under light irradiation, the encoded VEGFR2 gene was released and expressed in tumor tissues, which can suppress angiogenesis induced by a T cell-mediated autoimmune response and inhibit tumor growth.
- This article is part of the themed collections: Celebrating International Women’s Day: Women in Materials Science and 2021 Materials Horizons Advisory Board collection
 Please wait while we load your content...
Please wait while we load your content...

