
Synergy between dynamic covalent boronic ester and boron–nitrogen coordination: strategy for self-healing polyurethane elastomers at room temperature with unprecedented mechanical properties†

Abstract
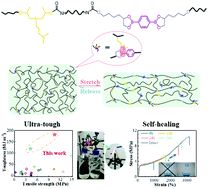
Achieving mechanical robustness and highly efficient self-healing simultaneously at room temperature is always a formidable challenge for polymeric materials. Herein, a series of novel supramolecular polyurethane elastomers (SPUEs) are developed by incorporating dynamic covalent boronic ester and boron–nitrogen (B–N) coordination. The SPUEs demonstrate the highest tensile toughness (∼182.2 MJ m−3) to date for room-temperature self-healable polymers, as well as an excellent ultimate tensile strength (∼10.5 MPa) and ultra-high fracture energy (∼72 100 J m−2), respectively, owing to a synergetic quadruple dynamic mechanism. It is revealed that the B–N coordination not only facilitates the formation and dissociation of boronic ester at room temperature but also dramatically enhances the mechanical properties by the intermolecular coordinated chain crosslinking and intramolecular coordinated chain folding. Meanwhile, the B–N coordination and urethane hydrogen interaction also serve as sacrificial bonds, which rupture during stretching to dissipate energy and recover after release, leading to superior notch insensitiveness and recoverability. The SPUEs restore their mechanical robustness after self-healing at room temperature and the self-healing efficiency can be dramatically accelerated by surface wetting.
- This article is part of the themed collections: Materials Horizons 10th anniversary regional spotlight collection: China, Materials Horizons Lunar New Year collection 2022 and 2021 Materials Horizons most popular articles
 Please wait while we load your content...
Please wait while we load your content...

