
Emerging investigator series: emerging disinfection by-product quantification method for wastewater reuse: trace level assessment using tandem mass spectrometry†

Abstract
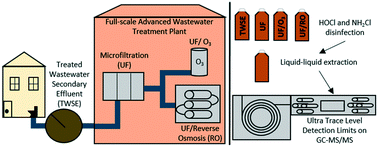
The availability of freshwater sources is declining as a result of increasing populations, economic activities, and climate change. These increasing trends will also drive up the demand for potable water that will require the use of alternative sources including wastewater-impacted and saline waters. Therefore, it is crucial to understand the formation of emerging toxic DBPs from advanced treatment of treated secondary wastewater effluents for potable reuse. In this study, a highly sensitive analytical method was developed to characterize 25 DBPs from 5 chemical classes (haloacetonitriles, halonitromethanes, haloacetaldehydes, haloketones, and iodinated trihalomethanes) in recycled wastewaters using a gas chromatography tandem mass spectrometer (MS/MS). The high sensitivity of MS/MS technology permitted a reduced sample concentration factor (50×) that required only 30 min of extraction time and 10 mL of sample volume. Method detection limits are the lowest reported between 2.0–68.9 ng L−1. Matrix effects in secondary wastewater effluents were low (0–30%) compared to ultra pure water. A full-scale facility for wastewater reuse that treated secondary wastewater effluents through microfiltration (UF), followed by ozone (UF/O3) or reverse osmosis (UF/RO) was evaluated. Water samples from each process were chlorinated (HOCl) and chloraminated (NH2Cl) to evaluate DBP precursor removal and DBP formation potential, the first study of its kind. Overall, HOCl formed higher summed DBP levels (0.5–18.5 μg L−1) compared to NH2Cl (0.2–8.5 μg L−1). HAN was significantly lower in UF/O3/HOCl (59%) and UF/RO/HOCl (99%) compared to UF/HOCl. However, HNM was enhanced after UF/O3/HOCl. In chloraminated samples, UF/O3/NH2Cl produced a higher amount of DBPs compared to UF/NH2Cl including haloacetonitriles, halonitromethanes, haloketones, and iodinated trihalomethanes.
- This article is part of the themed collections: Emerging Investigator Series and ENQA - 20th Brazilian Meeting on Analytical Chemistry
 Please wait while we load your content...
Please wait while we load your content...

