
New frontiers: 1,4-diphosphinines and P-bridged bis(NHCs)

Abstract
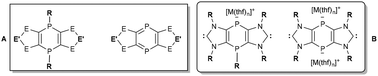
This review describes synthetic concepts and breakthroughs in 1,4-diphosphinine and related P-bridged bis(NHCs) chemistry, covering the last four decades, starting from monocyclic 1,4-dihydro-1,4-diphosphinines in the early 80s to the most recent and promising achievements of tricyclic 1,4-dihydro-1,4-diphosphinines and tricyclic 1,4-diphosphinines. Theoretical aspects are presented for 1,4-dihydro- and 1,4-diphosphinines considering HOMO LUMO situations as well as the degree of aromaticity. Moreover, fundamental characteristics of analytical data of these compounds are highlighted with special focus on substituent effects, structural aspects and trends of electrophilicity. The two P-centers and the heterocyclic rings of 1,4-dihydro- and 1,4-diphosphinines constitute a broad platform for substitution, reduction/oxidation, alkylation, complexation and cycloaddition reactions, i.e., a comprehensive compilation of reactivity aspects is presented. Furthermore, very recent developments in the synthesis and reactivity of tricyclic PV/V- and PIII/III-bridged bis(imidazole-2-ylidenes) will be discussed together with new perspectives derived from an antiaromatic middle ring. In total, our intention is to show new frontiers, i.e., new synthetic paths, thus creating novel opportunities for potential applications in molecular and materials chemistry.
- This article is part of the themed collection: 2021 Frontier and Perspective articles
 Please wait while we load your content...
Please wait while we load your content...

