
Virtual coformer screening by a combined machine learning and physics-based approach†

Abstract
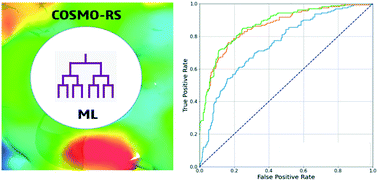
Cocrystals as a solid form technology for improving physicochemical properties have gained increasing popularity in the pharmaceutical, nutraceutical, and agrochemical industries. However, the list of potential coformers contains hundreds of molecules; far more than can be routinely screened and confirmed. Cocrystal screening experiments require significant amounts of active ingredients at an early project stage, and are expensive and time-consuming. Physics-based models and machine learning (ML) models have both been used to perform virtual cocrystal screening to guide experimental screening efforts, but both have certain limitations. Here, we present a combined ML/COSMO-RS fast virtual cocrystal screening method that proves to be significantly better than the sum of its parts in application to internal and external validation sets. To achieve that, we have defined the optimal threshold values of ML cocrystallization probability and COSMO-RS excess enthalpy of drug/coformer mixing for the combined coformer ranking. An approach to determine an applicability domain (AD) of the ML model has been implemented. The speed and accuracy of the new combined model allow it to be a good alternative to the physics-based CSP-based approach to support pharmaceutical projects with tight timeline and budget constraints.
- This article is part of the themed collections: Data Driven Crystal Engineering and Computer Aided Solid Form Design
 Please wait while we load your content...
Please wait while we load your content...

