
Electrides: a review

Abstract
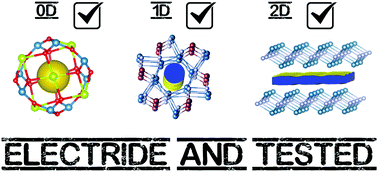
Electrides are systems in which an electron is not bound to an atom and plays an active role in the materials structure. We present concise summaries of the different types of electride in the literature, in which we systematically revisit their history and discuss their unique behaviour. We go on to offer intuitive guidelines for novel electride discovery and suggest advice for the accurate treatment of their physical properties. In the end, we conclude on the common nature of all electrides, with an alternative definition intended to minimise confusion surrounding these materials.
- This article is part of the themed collections: Journal of Materials Chemistry C Emerging Investigators, Journal of Materials Chemistry C Lunar New Year collection 2021 and 2020 Journal of Materials Chemistry C most popular articles
 Please wait while we load your content...
Please wait while we load your content...

