
Liquid metal-supported synthesis of cupric oxide†

Abstract
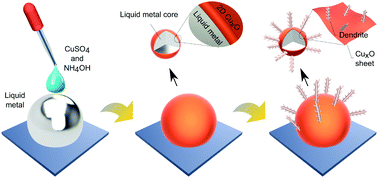
Liquid metal interfaces with their unique chemistry can provide an extraordinary tool to synthesise materials with controlled characteristics at room temperature. Here, the templated synthesis of cupric oxide (CuO) onto a gallium based liquid metal (galinstan) surface is explored. The interaction between galinstan and copper sulphate solution yields initial interfacial CuO sheets and then secondary dendritic nanostructures are grown from the sheets. It is shown that the pH of the aqueous environment plays a critical role in tuning the synthesis of substoichiometric CuxO to CuO. For enhancing the interfacial area and establishing heterojunctions, the liquid metal is processed into a suspension of micro and nano droplets by sonication and the example of using the synthesised nano structures for enhanced photocatalysis is demonstrated for the catalysis of a model dye. This study presents a facile approach for synthesising highly functional systems based on liquid metals, and it can be extended to explore other metal oxide–liquid metal composites.
- This article is part of the themed collections: 2020 Journal of Materials Chemistry C most popular articles and Celebrating our 2020 Prize and Award winners
 Please wait while we load your content...
Please wait while we load your content...

