
Advances and challenges in metallic nanomaterial synthesis and antibacterial applications
 a
and
Xuemei
Wang
*a
a
and
Xuemei
Wang
*a
Abstract
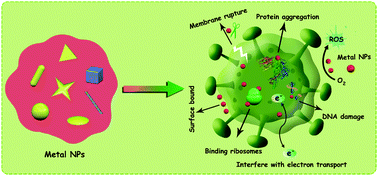
Multi-drug resistant bacterial infection has become one of the most serious threats to global public health. The preparation and application of new antibacterial materials are of great significance for solving the infection problem of bacteria, especially multi-drug resistant bacteria. The exceptional antibacterial effects of metal nanoparticles based on their unique physical and chemical properties make such systems ideal for application as antibacterial drug carriers or self-modified therapeutic agents both in vitro and in vivo. Metal nanoparticles also have admirable clinical application prospects due to their broad antibacterial spectrum, various antibacterial mechanisms and excellent biocompatibility. Nevertheless, the in vivo structural stability, long-term safety and cytotoxicity of the surface modification of metal nanoparticles have yet to be further explored and improved in subsequent studies. Herein, we summarized the research progress concerning the mechanism of metal nanomaterials in terms of antibacterial activity together with the preparation of metal nanostructures. Based on these observations, we also give a brief discussion on the current problems and future developments of metal nanoparticles for antibacterial applications.
- This article is part of the themed collections: 2020 Journal of Materials Chemistry B most popular articles and Hybrid Nanoparticle Composites
 Please wait while we load your content...
Please wait while we load your content...

