
AromTool: predicting aromatic stacking energy using an atomic neural network model†
Ruibo
Wu  *a
*a
*a
Abstract
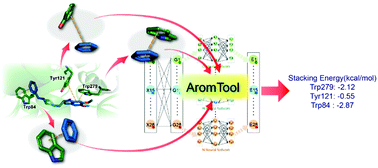
Aromatic stacking exists widely and plays important roles in protein–ligand interactions. Computational tools to automatically analyze the geometry and accurately calculate the energy of stacking interactions are desired for structure-based drug design. Herein, we employed a Behler–Parrinello neural network (BPNN) to build predictive models for aromatic stacking interactions and further integrated it into an open-source Python package named AromTool for benzene-containing aromatic stacking analysis. Based on extensive testing, AromTool presents desirable precision in comparison to DFT calculations and excellent efficiency for high-throughput aromatic stacking analysis of protein–ligand complexes.
 Please wait while we load your content...
Please wait while we load your content...

