
Construction of all-carbon quaternary stereocenters by catalytic asymmetric conjugate addition to cyclic enones in natural product synthesis

Abstract
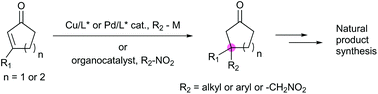
Asymmetric catalysis for chiral compound synthesis is a rapidly growing field in modern organic chemistry and provides enantioselective materials to meet the demands of various fields. However, the construction of all-carbon quaternary stereocenters poses a distinct challenge in organic synthesis. The development of catalytic asymmetric conjugate additions that require only a catalytic amount of a transition metal with a chiral ligand or organocatalyst has provided an efficient approach to the preparative-scale synthesis of enantioselective and/or diastereoselective conjugate adducts. Such reactions have been used in various synthetic applications such as natural product synthesis and reports of the use of this approach are becoming increasingly common in the literature. In particular, tandem copper-catalyzed asymmetric conjugate addition/enolate trapping by a carbon electrophile enables diastereoselective synthesis of α,β-substituted ketones with contiguous stereogenic centers, which is still an intricate task in organic synthesis. In this review, the use of asymmetric conjugate addition in natural product synthesis is described and discussed in depth.
 Please wait while we load your content...
Please wait while we load your content...

