
Transition metal-catalyzed coupling of heterocyclic alkenes via C–H functionalization: recent trends and applications

Abstract
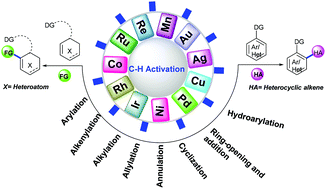
Heterocyclic alkenes represent an important class of reactive feedstock and valuable synthons for the synthesis of biologically important heterocyclic scaffolds. Although functionalized heterocyclic alkenes and their derivatives can be accessed via metal-catalyzed cross-coupling reactions, yet the prefunctionalisation of starting materials makes the process a bit cumbersome. On the other hand, transition-metal-catalyzed C–H functionalization is one of the most persuasive and front-line research areas in modern synthetic organic chemistry for atom/step-economy, viability and high efficacy. This critical review highlights the recent advances in coupling of heterocyclic alkenes by transition-metal-catalyzed reactions with special emphasis on arylation, alkenylation, alkylation, hydroarylation, ring-opening addition and annulation/cyclization via the C–H functionalization strategy reported since 2015.
 Please wait while we load your content...
Please wait while we load your content...

