
Tuning aggregation-induced emission nanoparticle properties under thin film formation†

Abstract
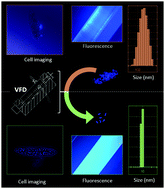
The most frequently used approach to preparing aggregation-induced emission fluorogen (AIEgen) particles is precipitation. Therefore, the addition of an AIEgen solution into water results in the formation of AIEgen particles in a very short time. Within such a short period of time and in the absence of proper mixing under shear, AIE particles are likely to be distributed in a wide range of sizes, thereby affecting their ultimate brightness and applications. Despite numerous attempts, the size of AIEgen particles is still within the range of 200–300 nm. For the first time, we developed a facile robust and cost-effective method for the fabrication of aggregation-induced emission nanoparticles with tuneable particle sizes <100 nm, high quantum yield, and excellent photostability. The direct diffusion of nanoparticles within the cell or in a single-celled organism, as an advantage of size reduction, opens new opportunities for biological and material studies. Such a significant reduction in AIE nanoparticle size has the potential for developing more efficient techniques for characterizing advanced nanomaterials and understanding biological processes and detection strategies.
- This article is part of the themed collection: Recent Progress on Aggregation-Induced Emission
 Please wait while we load your content...
Please wait while we load your content...

