
Recent advances in MOF-based photocatalysis: environmental remediation under visible light
 *b
*b
Abstract
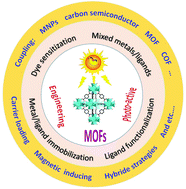
Visible light-induced photocatalysis is a promising way for environmental remediation due to efficient utilization of solar energy. Recently, metal–organic frameworks (MOFs) have attracted increasing attention in the field of photocatalysis. In comparison with traditional metal oxide semiconductors, MOFs have many advantages, such as high specific surface area, rich topology and easily tunable porous structure. In this review, we aim to summarize and illustrate recent advances in MOF-based photocatalysis for environmental remediation under visible light, including wastewater treatment, air purification and disinfection. A series of strategies have been designed to modify and regulate pristine MOFs for enhanced photocatalytic performance, such as ligand functionalization, mixed-metal/linker strategy, metal ion/ligand immobilization, dye sensitization, metal nanoparticle loading, carbon material decoration, semiconductor coupling, MOF/COF coupling, carrier loading and magnetic recycling. The above modifications may result in extended visible light absorption, efficient generation, separation and transfer of photogenerated charges, as well as good recyclability. However, there are still many challenges and obstacles. In order to meet the requirements of using MOF photocatalysis as a friendly and stable technology for low-cost practical applications, its future development prospects are also discussed.
 Please wait while we load your content...
Please wait while we load your content...

