
Dynamic covalent exchange in poly(thioether anhydrides)†

Abstract
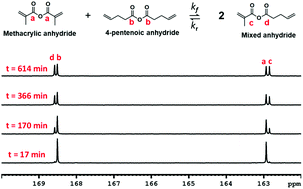
Dynamic covalent exchange (DCE) of anhydride moieties is examined in both model compounds and network polymers. In addition, the polyanhydrides are shown to be capable of self-healing and recycling. Specifically, thermodynamic and kinetic analysis of the exchange process in symmetric model compounds methacrylic anhydride and 4-pentenoic anhydride, which produce an asymmetric anhydride upon exchange, was undertaken using 13C NMR spectroscopy. The equilibrium constant (Keq) was determined to be approximately 2–3, and the activation energy (Ea) for the production of the asymmetric anhydride was 178 kJ mol−1. The rate of exchange was shown to be increased catalytically in the presence of carboxylic acid. Poly(thioether anhydrides) were made using 4-pentenoic anhydride and mixtures of dithiol (1,6-hexanedithiol) and tetrathiol (pentaerythritol tetrakis(3-mercaptopropionate)) via radical-mediated thiol–ene polymerizations, and network relaxation rates studied using dynamic mechanical analysis. The activation energy and stress relaxation times for various polymer compositions were correlated to crosslink density and polymer composition. It was determined that cross-link density of the polymer system does not significantly impact Ea; however, relaxation time (τ) nearly quadrupled from the least cross-linked system to the most cross-linked system. The network polymers exhibited self-healing and facile recycling behavior, which we attributed to the anhydride DCE process that occurs readily in these materials at elevated temperatures. Using the composition with the fastest relaxation, we observed complete visible recovery of damage (notch) within a 4 hours duration at 90 °C. Recycling under compression molding for 15 minutes using 76 MPa hydrostatic pressure at 90 °C was successful. Both healed and recycled samples were successful in recovering or surpassing their original mechanical properties.
- This article is part of the themed collection: Polymer Chemistry Pioneering Investigators 2021
 Please wait while we load your content...
Please wait while we load your content...

