
Single-chain crosslinked polymers via the transesterification of folded polymers: from efficient synthesis to crystallinity control†

Abstract
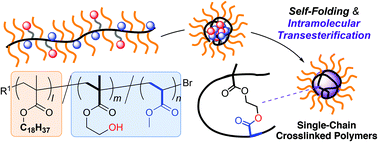
Herein we report efficient synthetic systems of single-chain crosslinked polymers via the intramolecular transesterification of folded random copolymers in organic media and the unique crystallization behavior of their crosslinked polymers. For this purpose, we designed random terpolymers comprising octadecyl methacrylate (ODMA), 2-hydroxyethyl methacrylate, and methyl acrylate (MA). The copolymers self-folded in octane via the association of the hydroxyl groups to form reverse micelles; the micelles were intramolecularly crosslinked by scandium-mediated transesterification of the MA units with the hydroxyl groups to give polymer nanoparticles bearing multiple octadecyl groups. This system affords efficient synthesis of various single-chain crosslinked polymers with controlled molecular weight at a relatively high concentration of up to 50 mg mL−1. Additionally, the crystallinity of these ODMA-based copolymers can be controlled by the crosslinking degree; the crystallinity and melting temperatures of the octadecyl groups gradually decreased as the intramolecular crosslinking of the copolymers proceeded. Thus, controlling the intramolecular crosslinking of polymers is one possibility to tune the crystallinity of polymer materials.
 Please wait while we load your content...
Please wait while we load your content...

