
Nanostructured potassium–organic framework as an effective anode for potassium-ion batteries with a long cycle life†

Abstract
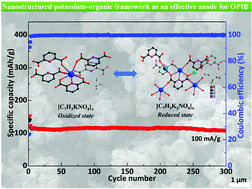
Finding new organic materials to address several issues (e.g. capacity, stability, and cycle life) in organic potassium-ion batteries (OPIBs) is very important and highly desirable. Here, to directly investigate the redox reaction of organic pyridine dicarboxylate in OPIBs and to avoid the interference from the redox-active metal ions, a non-redox-metal potassium metal–organic framework (K-MOF), [C7H3KNO4]n, based on pyridine-2,6-dicarboxylic acid (H2PDA), has been successfully synthesized and applied as a promising organic anode for long-cycle life PIBs. The crystal structure of [C7H3KNO4]n was confirmed by single-crystal X-ray diffraction analysis and FT-IR spectra. Moreover, the potassium-storage mechanism of organic pyridine dicarboxylate ligand was revealed by ex situ FT-IR/XRD characterization and theoretical calculations. The as-synthesized K-MOF resulted in a unique and reversible three-step redox reaction, exhibited superior electrochemical performance with the aid of N–K/O–K coordination bonds, and showed a high average specific capacity of 115 mA h g−1 at 100 mA g−1 for 300 cycles with the capacity retention of 92%.
- This article is part of the themed collections: Editor’s Choice: Functional MOFs and COFs, Battery science and technology – powered by chemistry, Advanced Nanomaterials for Energy Conversion and Storage and Nanoscale Most Popular 2020 Articles
 Please wait while we load your content...
Please wait while we load your content...

