
Gold nanonails for surface-enhanced infrared absorption†
 a
Jianfang
Wang
*b
a
Jianfang
Wang
*b
Abstract
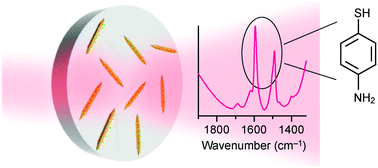
Surface-enhanced infrared absorption (SEIRA) can dramatically enhance the vibrational signals of analyte molecules owing to the interaction between plasmons and molecular vibrations. It has huge potential for applications in various detection and diagnostic fields. High-aspect-ratio rod-like metal nanostructures have been the most widely studied nanomaterials for SEIRA. However, nearly all of the rod-like nanostructures reported previously are fabricated using physical methods. They suffer from damping and low areal number densities. In this work, high-aspect-ratio Au nanorods are synthesized, and Au nanonails are prepared through Au overgrowth on the as-prepared Au nanorods. The aspect ratios of the Au nanorods and nanonails can be varied in the range of ∼10 to ∼60, and their longitudinal dipolar plasmon resonance wavelengths can be correspondingly tailored from ∼1.6 to ∼8.3 μm. The Au nanonails exhibit superior SEIRA performance with 4-aminothiophenol used as the probe molecules. They are further used to detect the common biomolecule L-cysteine. Numerical simulations are further performed to understand the experimental results. They match well with the experimental observations, revealing the mechanism of the SEIRA enhancement. Our study demonstrates that colloidal high-aspect-ratio Au nanonails and nanorods can function as SEIRA nanoantennas for highly sensitive molecular detection in various situations.
- This article is part of the themed collections: Nanoscale Horizons 10th anniversary regional spotlight collection: China and Horizons Community Board Collection: Optical and Photonic Materials
 Please wait while we load your content...
Please wait while we load your content...

