
Implementation of ferroelectric materials in photocatalytic and photoelectrochemical water splitting

Abstract
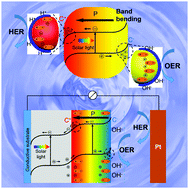
As a promising technology for sustainable hydrogen generation, photocatalytic (PC) and photoelectrochemical (PEC) water splitting have gathered immense attention over a half-century. While many review articles have covered extensive research achievements and technology innovations in water splitting, this article focuses on illustrating how the ferroelectric polarization influences charge separation and transportation in photocatalyst heterostructures during PC and PEC water splitting. This article first discusses the fundamentals of PC and PEC water splitting and how these electrochemical processes interact with the ferroelectric polarization-induced interfacial band bending, known as piezotronics. A few representative ferroelectric material-based heterogeneous photocatalyst systems are then discussed in detail to illustrate the effects of polarization, space charge region, and free charge concentration, which are critical factors determining the ferroelectric influences. Finally, a forward looking statement is provided to point out the research challenges and opportunities in this promising interdisciplinary research field between ferroelectrics and electrochemistry for clean energy applications.
- This article is part of the themed collection: Horizons Community Board Collection: Solar Energy Conversion
 Please wait while we load your content...
Please wait while we load your content...

