
Recent developments in polydopamine fluorescent nanomaterials

Abstract
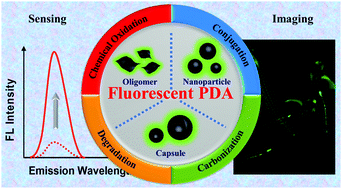
Polydopamine (PDA) fluorescent materials have recently gained much attention due to their unique physicochemical and biological properties. Since the first report on fluorescent PDA nanoparticles, many efforts have been made to develop new fabrication strategies to achieve their desirable luminescence properties. This feature article summarizes the recent advances and current highlights in fluorescent PDA materials including fabrication methods, nanomaterial morphologies, luminescence mechanisms and applications for imaging and sensing in the last few years. We believe that this review will provide new insights into the mechanistic aspect of PDA fluorescence for guiding preparation approaches and new applications.
- This article is part of the themed collection: Horizons Community Board Collection: Optical and Photonic Materials
 Please wait while we load your content...
Please wait while we load your content...

