
A high-throughput method to characterize the gut bacteria growth upon engineered nanomaterial treatment†

Abstract
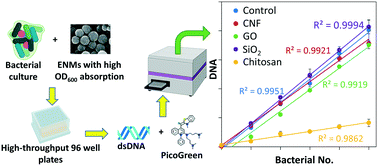
Humans are increasingly exposed to various types of engineered nanomaterials (ENMs) via dietary ingestion of nano-enabled food products, but the impact of these ENMs on the gut bacteria health is still poorly understood. Current efforts in understanding the impact of these ENMs are hampered by their optical interferences in conventional quantification and viability assays, such as optical density and whole cell fluorescence staining assays. Therefore, there is a need to develop a more reliable bacteria quantification method in the presence of ENMs to effectively screen the potential adverse effects arising from the exposure to increasing ENMs on the human gut microbiome. In this study, we developed a DNA-based quantification (DBQ) method in a 96-well plate format. A post-spiking method was used to correct the interference from ENMs in the reading. We showed the applicability of this method for several types of ENMs, i.e., cellulose nanofibers (CNFs), graphene oxide (GO), silicon dioxide (SiO2), and chitosan, both in pure bacterial culture and in vitro human gut microbiome community. The detection limit for the highest dosing of CNF, GO, SiO2, and chitosan ENMs was approximately 0.18, 0.19, 0.05, and 0.24 as OD600, respectively. The method was also validated by a dose response experiment of E. coli with chitosan over the course of 8 h. We believe that this method has great potential to be used in screening the effect of ENMs on the growth of gut bacteria or any other in vitro models and normalization for metabolite or protein analysis.
- This article is part of the themed collections: Nano-bio interactions and Research from our newest ESPI Editorial Board members
 Please wait while we load your content...
Please wait while we load your content...

