
The phosphinoboration of acyl chlorides†

Abstract
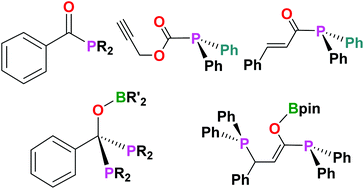
This investigation examines the reactivity of phosphinoboronate esters Ph2PBpin (pin = 1,2-O2C2Me4) and Ph2PBcat (cat = 1,2-O2C6H4), as well as other phosphinoboron species, with various aryl and aliphatic acyl chlorides. These reactions proceed smoothly to give acyl phosphines of the type RC(O)PR′2 along with loss of a boron-chloride compound. In some cases, a second equivalent of the phosphinoboron species can add to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bond at elevated temperatures to give the corresponding diphosphines RC(OBR′′2)(PR′2)2. These ambiphilic diphosphines behave like substituted (1,1-bis(diphenylphosphino)methane) derivatives in a reaction of PhC(OBpin)(PPh2)2 (2a) with (η5-C9H7)Rh(η2-coe)2 (coe = cis-cyclooctene) affording the indenyl rhodium complex (η5-C9H7)Rh(PhC(OBpin)(PPh2)2) (3a) where the phosphines are bound to the metal centre in a κ2-P,P bidentate manner.
O double bond at elevated temperatures to give the corresponding diphosphines RC(OBR′′2)(PR′2)2. These ambiphilic diphosphines behave like substituted (1,1-bis(diphenylphosphino)methane) derivatives in a reaction of PhC(OBpin)(PPh2)2 (2a) with (η5-C9H7)Rh(η2-coe)2 (coe = cis-cyclooctene) affording the indenyl rhodium complex (η5-C9H7)Rh(PhC(OBpin)(PPh2)2) (3a) where the phosphines are bound to the metal centre in a κ2-P,P bidentate manner.
- This article is part of the themed collections: Celebrating our Golden Authors and Celebrating our 2021 Prizewinners
 Please wait while we load your content...
Please wait while we load your content...

