
Rational design and latest advances of polysaccharide-based hydrogels for wound healing
 *a
and
Fu-Jian
Xu
*b
*a
and
Fu-Jian
Xu
*b
Abstract
Acute and chronic wounds cause severe physical trauma to patients and also bring an immense socio-economic burden. Hydrogels are considered to be effective wound dressings. Polysaccharides possessing distinctive properties such as biocompatibility, biodegradability, and nontoxicity are promising candidates to structure hydrogels for wound healing. Polysaccharide-based hydrogels can provide suitable moisture for the wound and act as a shield against bacteria. Adequate mechanical properties, degradability, and therapeutic agent controlled release of polysaccharide-based hydrogels have been already characterized for effective utilization. This review presented several crucial design considerations about hydrogels for wound healing, and the current state of polysaccharide (chitosan, alginate, hyaluronic acid, cellulose, dextran, and starch)-based hydrogels as wound dressings was also summarized. The commonly used crosslinking techniques, including physical, chemical, and enzymatic crosslinking, are discussed in detail. Finally, we outline the challenges and perspectives about the improvement of polysaccharide-based hydrogels.
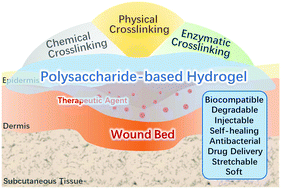
- This article is part of the themed collections: Biomaterials Science 10th Anniversary: Top papers from China and Biomaterials Science Most Popular 2020
 Please wait while we load your content...
Please wait while we load your content...

