
Enzyme-inspired flavin–polydopamine as a biocompatible nanoparticle photocatalyst†

Abstract
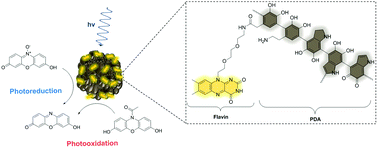
A new approach aimed at designing an enzyme-inspired photocatalyst is presented that exploits the inherent photocatalytic activity of flavin and the facile polymerization of dopamine to afford hybrid cofactor-containing nanoparticles. The flavin–polydopamine system benefits from ease of synthesis, tunability in terms of size and activity, and excellent temporal control over the catalyzed reactions. This novel, versatile photocatalyst exhibits both photooxidation and photoreduction of chromogenic enzymatic substrates. In addition, the prepared hybrid nanoparticles are shown to be non-toxic, paving the way to their use in a wider range of applications beyond green catalysis, such as antioxidant adjuvants to various therapeutic approaches.
- This article is part of the themed collections: Emerging horizons in polymer applications and Horizons Community Board Collection: Solar Energy Conversion
 Please wait while we load your content...
Please wait while we load your content...

