
The interparticle distance limit for multiple exciton dissociation in PbS quantum dot solid films†

Abstract
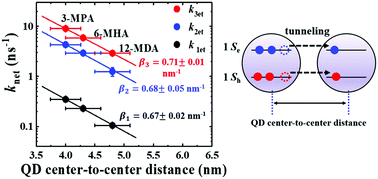
Understanding the behaviour of multiple exciton dissociation in quantum dot (QD) solid films is of fundamental interest and paramount importance for improving the performance of quantum dot solar cells (QDSCs). Unfortunately, the charge transfer behaviour of photogenerated multiple exciton in QD solid films is not clear to date. Herein, we systematically investigate the multiple exciton charge transfer behaviour in PbS QD solid films by using ultrafast transient absorption spectroscopy. We observe that the multiple exciton charge transfer rate within QD ensembles is exponentially enhanced as the interparticle distance between the QDs decreases. Biexciton and triexciton dissociation between adjacent QDs occurs via a charge transfer tunneling effect just like single exciton, and the charge tunneling constants of the single exciton (β1: 0.67 ± 0.02 nm−1), biexciton (β2: 0.68 ± 0.05 nm−1) and triexciton (β3: 0.71 ± 0.01 nm−1) are obtained. More importantly, for the first time, the interparticle distance limit (≤4.3 nm) for multiple exciton charge transfer between adjacent QDs is found for the extraction of multiple excitons rapidly before the occurrence of Auger recombination. This result points out a vital and necessary condition for the use of multiple excitons produced in PbS QD films, especially for their applications in QDSCs.
 Please wait while we load your content...
Please wait while we load your content...

