
Mineralization of magnetic nano-tape in self-organized nanospace composed of nucleopeptides and peptides†
 *a
*a
Abstract
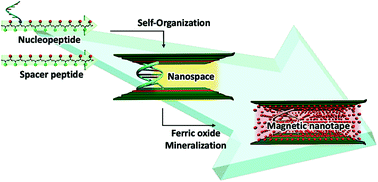
Self-organization based on specific interactions has been actively studied for the formation of highly ordered and hierarchical structures on the molecular scale. We designed and synthesized a nucleopeptide, Ac-VEVS(g(GC)3)(VE)7-CONH2, having a nucleotide graft-chain in a peptide main-chain, and a spacer peptide acting as a template for ferric oxide mineralization. The synthesized nucleopeptide and spacer peptide mixture formed a self-organized assembly having a nanospace owing to the complementary base pairing between the nucleotide graft-chains of the nucleopeptides and intermolecular hydrogen-bonding between the peptide chains on the substrates. In this study, we report the selective mineralization of magnetite in the nanospace which acted as a reaction field. Mineralized magnetite formed an oriented nano-tape, whose morphology was similar to that of the template nanosheet. This implies that the nanospace in the ordered nanosheet formed by self-organization processes of the nucleopeptide and spacer peptide is extremely useful as a reaction field for the mineralization of structure-controlled magnetite.
 Please wait while we load your content...
Please wait while we load your content...

