
Rapid immunodiagnostics of multiple viral infections in an acoustic microstreaming device with serum and saliva samples

Abstract
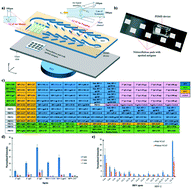
There is a growing need to screen multiple infections simultaneously rather than diagnosis of one pathogen at a time in order to improve the quality of healthcare while saving initial screening time and reduce costs. This is the first demonstration of a five-step protein array assay for the multiplexed detection of HIV, HPV and HSV antibodies on an integrated microfluidic system. HIV, HPV and HSV reactive antibodies from both serum and saliva were rapidly detected by acoustic streaming-based mixing and pumping to enable an integrated, rapid and simple-to-use multiplexed assay device. We validated this device with 37 serum and saliva samples to verify reactivity of patient antibodies with HIV, HPV and HSV antigens. Our technology can be adapted with different protein microarrays to detect a variety of other infections, thus demonstrating a powerful platform to detect multiple putative protein biomarkers for rapid detection of infectious diseases. This integrated microfluidic protein array platform is the basis of a potent strategy to delay progression of primary infection, reduce the risk of co-infections and prevent onward transmission of infections by point-of-care detection of multiple pathogens in both serum and oral fluids.
- This article is part of the themed collection: Coronavirus articles - free to access collection
 Please wait while we load your content...
Please wait while we load your content...

