
Preferential proton conduction along a three-dimensional dopant network in yttrium-doped barium zirconate: a first-principles study†
 *a
Donglin
Han
a
and
*a
Donglin
Han
a
and
Abstract
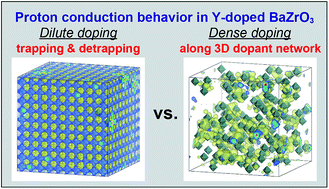
The atomic-scale picture of the proton conduction in Y-doped BaZrO3 has theoretically been investigated using first-principles calculations on the basis of the nudged elastic band (NEB) method and the kinetic Monte Carlo (KMC) method. In this crystal, protons mainly reside around Y dopants due to the electrostatic attractive interaction between the dopants and protons, which is well known as proton trapping. In the case of the typical doping level x ∼ 0.2 in BaZr1−xYxO3−δ, the existence of Y–Y–Y triplets with a triangular configuration is an origin of the strong proton-trapping effect, in which protons are transferred in an oscillatory manner between two adjacent sites. The proton conduction behavior is however different from the conventional mechanism of trapping & detrapping applicable only to the case of dilute doping. At the typical doping level with dense dopants, protons preferentially migrate along the three-dimensional network of Y dopants throughout the crystal without detrapping. The preferential conduction pathways moderate the strong trapping effect of dense dopants, resulting in a minor reduction of the proton diffusivity and mobility in highly doped BaZrO3.
 Please wait while we load your content...
Please wait while we load your content...

