
Boron-doped coronenes with high redox potential for organic positive electrodes in lithium-ion batteries: a first-principles density functional theory modeling study†
 *b
and
*b
and
Abstract
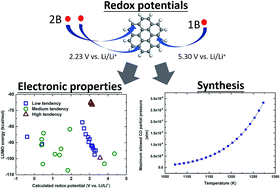
Boron-doped coronenes are attractive as promising positive electrode materials for lithium-ion batteries due to the unique physical and chemical properties of coronene. In this study, we computationally investigate the thermodynamics and redox properties of boron-doped coronenes using the first-principles density functional theory method to evaluate their potential as positive electrodes. It is found from our computations that the redox potentials of the boron-doped coronenes change as a function of the number of doped boron atoms and their distribution, predicting that the weight-averaged redox potentials of one-boron- and two-boron-doped coronenes would reach 5.30 V and 2.23 V, respectively, with respect to Li/Li+. It is highlighted that the highest redox potential of 5.42 V vs. Li/Li+ is predicted for the coronene with a boron atom doped at an edge position. Further investigation on the electronic properties of the boron-doped coronenes provides a strong correlation of the redox potential with the electron affinity, but poor correlation with the lowest unoccupied molecular orbital. Last, a proposed synthetic route that utilizes a pristine coronene reacting with B2O3 and Mg for developing boron-doped-coronenes provides viable thermodynamic conditions of the released CO pressures of ∼1.3 × 10−6 atm at 1073 K and ∼2.8 × 10−5 atm at 1273 K.
- This article is part of the themed collection: Materials and Nano Research in Atlanta
 Please wait while we load your content...
Please wait while we load your content...

