
Liquid crystal gelators with photo-responsive and AIE properties†

Abstract
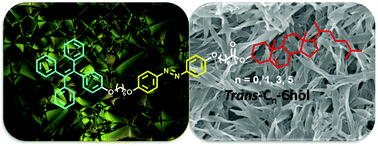
A series of new liquid crystal (LC) gelators with photo-responsive and AIE properties were synthesized by connecting cholesterol and tetraphenylethylene (TPE) to a central azobenzene moiety (trans-Cn-Chol, n = 0, 1, 3, 5), in which alkyl chains with different lengths (CH2)n were used as spacer to adjust the distance between cholesterol and azobenzene, while a fixed alkyl chain (CH2)6 was placed between azobenzene and TPE. All these trans-Cn-Chol molecules exhibit LC phase of type smectic A. Three derivatives of trans-Cn-Chol (with n = 0, 1, 3) keep their smectic A LC phases at room temperature upon cooling after at least one heating history to isotropic (Iso) phase, and present hot-crystallization (94–104 °C) upon further heating. Moreover, their thin films with LC state at room temperature can undergo photo-responsive LC–Iso phase transition under UV irradiation because of the trans–cis photo-isomerization of azobenzene. On the other hand, as gelators trans-Cn-Chol with long spacers, trans-C3-Chol and trans-C5-Chol, form stable low-molecular weight organogels (LMOGs) in various solvents, whereas trans-C0-Chol and trans-C1-Chol with short spacers cannot form stable LMOGs in tested solvents. Diverse supramolecular chiral structures like “sea urchins”, helical fibers, “honeycomb” and twisted ribbons were observed for both trans-C3-Chol and trans-C5-Chol gels. On account of the involvement of TPE and azobenzene moieties, these molecules present significant aggregation-induced emission (AIE) and gel-enhanced emission, and their LMOGs exhibit photo-responsive gel–sol transition. These LC molecules with gelation ability, AIE characteristics and photo-responsive properties can have potential applications as sensors and optoelectronic materials.
- This article is part of the themed collection: Recent Progress on Aggregation-Induced Emission
 Please wait while we load your content...
Please wait while we load your content...

