
pH-Responsive aggregation-induced emission of Au nanoclusters and crystallization of the Au(i)–thiolate shell†
 *a
and
*a
and
Abstract
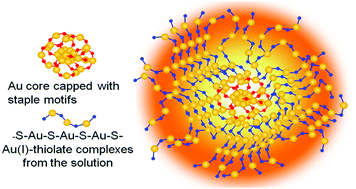
Aggregation-induced emission (AIE) is an attractive strategy for enhancing the luminescence of metal nanoclusters (NCs). However, rational control of the AIE of Au NCs is currently one of the key challenges and fundamental understanding of the complex surface/interfacial structures, and the emission mechanism of AIE-type Au NCs is far from complete. In this study, we report a pH-controllable AIE accompanied with a self-assembly/disassembly process of Au NCs with Au(I)–thiolate complexes with glutathione (GSH)-protected AIE-type Au NCs as a model. It was found that the solution pH could be used to control the landscape of the Au(I)–thiolate complexes encapsulating the Au NCs, generating varied AIE. Transmission electron microscopy (TEM) studies revealed that the Au NCs were essentially embedded inside the pH-sensitive supramolecular assemblies of the Au(I)–thiolate complexes. It was further found that a certain degree of crystallization of the Au(I)–thiolate shell occurred on the surface of the Au NCs. This study expands the knowledge of both the surface/interfacial structures of AIE-type metal NCs and the control of their PL and their self-assembly.
- This article is part of the themed collection: Recent Progress on Aggregation-Induced Emission
 Please wait while we load your content...
Please wait while we load your content...

