
Label-free in-flow detection of receptor recognition motifs on the biomolecular corona of nanoparticles†
 a
H.
Lopez,
b
a
H.
Lopez,
b
Abstract
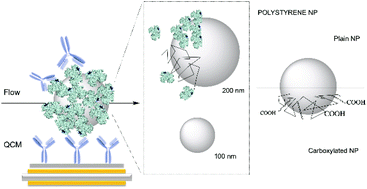
Nanomedicine, nanotargeting and nanotherapeutics have in the last few years faced several difficulties in translating the promising results obtained in vitro to an in vivo scenario. The origin of this discrepancy might be found in the lack of a detailed and realistic characterization of the biological surface of nanoparticles. Despite the capability to engineer nanomaterials with a great variety and a precise control of the surface functionalization, the targeting capability is lost when the nanoparticles are embedded in complex biological media, due to the formation of a biological layer (biomolecular corona). This biological layer represents the ultimate nanoparticle surface, likely to interact with the cell machinery. Therefore, in addition to traditional nanoparticle characterization techniques, a more insightful investigation of the biomolecular corona is needed, including the capability to assess the orientation and functionality of specific key molecular features. Here we present a method for the rapid screening of exposed protein recognition motifs on the surface of nanoparticles exploiting quartz crystal microbalance (QCM). We quantify accessible functional epitopes of transferrin-coated nanoparticles and correlate them to differences in nanoparticle size and functionalization. The target recognition occurs label free in flow, thereby pushing our investigation into a more in vivo-like scenario. Our method is applicable to a wide array of nanoparticles and therefore holds the potential to become an advanced technique for the classification of all kinds of nanobioconstructs based on their biological external functionality.
- This article is part of the themed collection: Editor’s Choice: Recent breakthroughs in nanobiotechnology
 Please wait while we load your content...
Please wait while we load your content...

