
Strengths of non-covalent interactions in hydrogen-bonded complexes B⋯HX and halogen-bonded complexes B⋯XY (X, Y = F, Cl): an ab initio investigation†
 *a
and
Anthony
Legon
*b
*a
and
Anthony
Legon
*b
Abstract
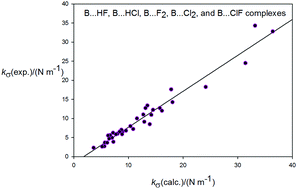
The intermolecular quadratic stretching force constants kcalc.σ of a series of hydrogen-bonded and halogen-bonded complexes B⋯HX and B⋯XY, where B is N2, CO, HCCH, C2H4, H2S. PH3, H2O or NH3 and X and Y are F or Cl, have been calculated ab initio at the CCSD(T)/aug-cc-pVTZ level of theory. The values obtained are compared with those kexp.σ values available from experimental centrifugal distortion constants by the use of a model based on the assumption of rigid, unperturbed component molecules at their equilibrium separation. Although the expressions resulting from the model involve equilibrium spectroscopic constants of B, HX (or XY), and B⋯HX (or XY), only zero-point quantities are normally available and are therefore used. A graph of kexp.σversus kcalc.σ for all complexes investigated can be fitted by linear regression as a straight line through the origin and with gradient 0.94(3), with a small scatter of points relative to the line. The scatter is attributed mainly to the use of zero-point spectroscopic constants in place of equilibrium values. Values of the equilibrium dissociation energy De(CBS) calculated at the CCSD(T)/CBS level (where CBS indicates complete basis set extrapolation using aug-cc-pVTZ and aug-cc-pVQZ basis sets) for all complexes are found to be directly proportional to kcalc.σ to a good degree of approximation, with a constant of proportionality obtained from the fit of the hydrogen-bonded complexes identical to that for halogen-bonded complexes within two standard deviations.
- This article is part of the themed collection: The halogen bond: a new avenue in recognition and self-assembly
 Please wait while we load your content...
Please wait while we load your content...

