
Evaluation of affibody charge modification identified by synthetic consensus design in molecular PET imaging of epidermal growth factor receptor†

Abstract
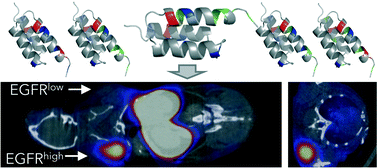
Tumor overexpression of epidermal growth factor receptor (EGFR) correlates to therapeutic response in select patient populations. Thus, molecular positron emission tomography (PET) imaging of EGFR could stratify responders versus non-responders. We previously demonstrated effectiveness of a “synthetic consensus” design principle to identify six neutralizing mutations within a 58-amino acid EGFR-targeted affibody domain. Herein, we extend the approach to identify additional neutralized variants that vary net charge from −2 to either −4 or +4 while retaining high affinity (1.6 ± 1.2 nM and 2.5 ± 0.7 nM), specific binding to EGFR, secondary structure, and stability (Tm = 68 °C and 59 °C). We radiolabeled the resultant collection of five charge variants with 64Cu and evaluated PET imaging performance in murine models with subcutaneously xenografted EGFRhigh and EGFRlow tumors. All variants exhibited good EGFRhigh tumor imaging as early as 1 h, with EA35S (+3/−5) achieving 7.7 ± 1.4 %ID g−1 tumor at 4 h with 1.5 ± 0.3 %ID g−1 EGFRlow tumor, 34 ± 5 tumor : muscle and 12 ± 3 tumor : blood ratios. The positively charged EA62S mutant (+6/−2) exhibited 2.2–3.3-fold higher liver signal than the other variants (p < 0.01). The EA68 variant with higher charge density was more stable to human and mouse serum than neutralized variants. In a comparison of radiometal chelators, 1,4,7-triazacyclononane,1-glutaric acid-4,7-acetic acid (NODAGA) exhibited superior physiological specificity to 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA). In total, these studies comparatively evaluated a set of EGFR-targeted affibodies varying in net charge and charge density, which revealed functional variations that are useful in engineering an ideal probe for translational studies.
- This article is part of the themed collection: MSDE Emerging Investigators 2018
 Please wait while we load your content...
Please wait while we load your content...

