
Framework vs. side-chain amphidynamic behaviour in oligo-(ethylene oxide) functionalised covalent-organic frameworks†
Demetrius A.
Vazquez-Molina,  a
Jose L.
Mendoza-Cortes,
*bcd
James K.
Harper
*a
and
Fernando J.
Uribe-Romo
*a
a
Jose L.
Mendoza-Cortes,
*bcd
James K.
Harper
*a
and
Fernando J.
Uribe-Romo
*a
a
Jose L.
Mendoza-Cortes,
*bcd
James K.
Harper
*a
and
Fernando J.
Uribe-Romo
*a
Abstract
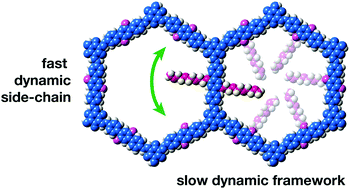
We present a family of covalent organic frameworks that have been functionalized with oligo-(ethylene oxide) chains of varying lengths. Because of the open structure of the COFs, the side chains do not interfere with their crystallization obtaining materials with predictable crystal structure. The difference in length of the side-chains allowed for the determination of amphidynamic behaviour with the use of 13C solid-state NMR relaxation methods. Computational calculations further contribute to understanding the atomistic dynamic behaviour of the different atoms. This study demonstrates the ability to design complex behaviour in organic crystals.
- This article is part of the themed collection: 2018 Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

