
Understanding the viral load during the synthesis and after rebinding of virus imprinted particles via real-time quantitative PCR†
 *a
*a
Abstract
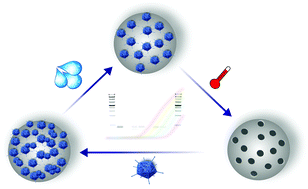
In the present study, virus imprinted particles have been synthesized for recognizing and specifically binding viruses. These materials may be used for biomimetic sensing schemes and for selective removal of virus particles. Virus imprinting procedures require careful optimization of the synthesis route for obtaining selective and efficiently binding imprinted materials. A remaining limitation has been a facile method for the quantification of the viral load during the imprinting process. Herein, human adenovirus (AdV) was selected as a model virus facilitating the development and application of a rapid virus quantification method based on a molecular biological approach. A real-time quantitative polymerase chain reaction, a.k.a., the qPCR method was developed for monitoring the AdV viral load during the synthesis of AdV imprinted particles, and subsequent rebinding studies. The developed analytical strategy allows the direct, rapid, and sensitive quantification of human adenovirus type 5 concentrations during synthesis and application of AdV imprinted polymers (AdV-MIPs) with a broad dynamic range suitable for both application scenarios. In addition, it was demonstrated by gel electrophoresis analysis that viruses indeed bind to the beads even after several washing steps.
- This article is part of the themed collection: Coronavirus articles - free to access collection
 Please wait while we load your content...
Please wait while we load your content...

