
Emerging investigator series: a 14-year depositional ice record of perfluoroalkyl substances in the High Arctic†

Abstract
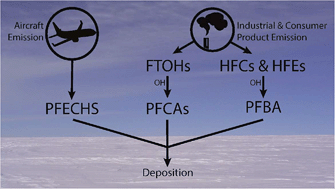
To improve understanding of long-range transport of perfluoroalkyl substances to the High Arctic, samples were collected from a snow pit on the Devon Ice Cap in spring 2008. Snow was analyzed for perfluoroalkyl acids (PFAAs), including perfluoroalkyl carboxylic acids (PFCAs) and perfluoroalkyl sulfonic acids (PFSAs), as well as perfluorooctane sulfonamide (FOSA). PFAAs were detected in all samples dated from 1993 to 2007. PFAA fluxes ranged from <1 to hundreds of ng per m2 per year. Flux ratios of even–odd PFCA homologues were mostly between 0.5 and 2, corresponding to molar ratios expected from atmospheric oxidation of fluorotelomer compounds. Concentrations of perfluorobutanoic acid (PFBA) were much higher than other PFCAs, suggesting PFBA loading on the Devon Ice Cap is influenced by additional sources, such as the oxidation of heat transfer fluids. All PFCA fluxes increased with time, while PFSA fluxes generally decreased with time. No correlations were observed between PFAAs and the marine aerosol tracer, sodium. Perfluoro-4-ethylcyclohexanesulfonate (PFECHS) was detected for the first time in an atmospherically – derived sample, and its presence may be attributed to aircraft hydraulic system leakage. Observations of PFAAs from these samples provide further evidence that atmospheric oxidation of volatile precursors is an important source of PFAAs to the Arctic environment.
- This article is part of the themed collections: Emerging Investigator Series, Research from our newest ESPI Editorial Board members and CSC100: Celebrating Canadian Chemistry
 Please wait while we load your content...
Please wait while we load your content...

