
Hydrogen-bond dynamics at the bio–water interface in hydrated proteins: a molecular-dynamics study†
Abstract
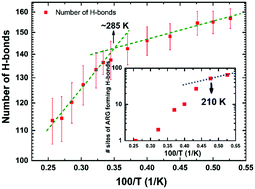
Water is fundamental to the biochemistry of enzymes. It is well known that without a minimum amount of water, enzymes are not biologically active. Bare minimal solvation for biological function corresponds to about a single layer of water covering enzymes' surfaces. Many contradictory studies on protein–hydration-water-coupled dynamics have been published in recent decades. Following prevailing wisdom, a dynamical crossover in hydration water (at around 220 K for hydrated lysozymes) can trigger larger-amplitude motions of the protein, activating, in turn, biological functions. Here, we present a molecular-dynamics-simulation study on a solvated model protein (hen egg-white lysozyme), in which we determine, inter alia, the relaxation dynamics of the hydrogen-bond network between the protein and its hydration water molecules on a residue-per-residue basis. Hydrogen-bond breakage/formation kinetics is rather heterogeneous in temperature dependence (due to the heterogeneity of the free-energy surface), and is driven by the magnitude of thermal motions of various different protein residues which provide enough thermal energy to overcome energy barriers to rupture their respective hydrogen bonds with water. In particular, arginine residues exhibit the highest number of such hydrogen bonds at low temperatures, losing almost completely such bonding above 230 K. This suggests that hydration water's dynamical crossover, observed experimentally for hydrated lysozymes at ∼220 K, lies not at the origin of the protein residues' larger-amplitude motions, but rather arises as a consequence thereof. This highlights the need for new experimental investigations, and new interpretations to link protein dynamics to functions, in the context of key interrelationships with the solvation layer.
 Please wait while we load your content...
Please wait while we load your content...

