
A “two-birds-one-stone” strategy to enhance capacitive deionization performance of flexible Ti3C2Tx MXene film electrodes by surface modification†
 *ab
*ab
Abstract
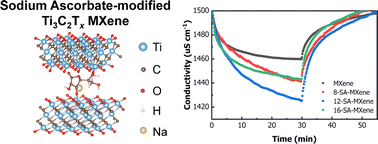
Two-dimensional transition metal carbides/nitrides (MXenes) have gained considerable prominence in capacitive deionization (CDI) due to their exceptional electrochemical activity and outstanding electronic conductivity. However, the further development of MXenes in practical applications in CDI is hampered by their limited narrow interlayer spacing and oxidation proneness. Herein, a dual-functional surface modification of Ti3C2Tx MXene by sodium ascorbate (SA) is proposed to concurrently enhance the salt adsorption capacity and long-term stability. The modification by SA induces synergistic functions, including the enlargement of interlayer spacing, effective protection of Ti from oxidation, and exceptional electrical conductivity. Simultaneously, this film electrode is designed to be flexible and free-standing, devoid of binders or adhesives, showing promise for the large-scale production of CDI electrodes. Benefiting from these advantages, the SA-modified MXene exhibit excellent desalination performance, including high salt adsorption capacity (109.6 mg g−1), high salt adsorption rates (17.5 mg g−1 min−1), and impressive cycling stability (100% retention after 80 cycles). And the adsorption behavior of SA-modified MXenes is further investigated by in situ X-ray diffraction and density functional theory calculations. This work proposes an effective modification and explores theoretical aspects for fabricating MXene-based electrodes suitable for CDI and other electrochemical applications in moist or aqueous environments.
- This article is part of the themed collections: 2024 Journal of Materials Chemistry A HOT Papers and Design and characterization of flexible electrode materials
 Please wait while we load your content...
Please wait while we load your content...

