
Optical study of electrochromic moving fronts for the investigation of ion transport in conducting polymers†
Abstract
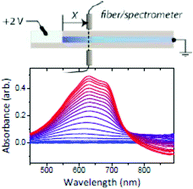
Conducting polymers, which can simultaneously support transport of ions in addition to electronic charges, form the basis for electrochemical devices that transduce ionic signals into electronic signals, and vice versa. Understanding and controlling mixed conduction is, however, challenging due to a lack of methodology to simultaneously probe ion (and hole) injection and/or transport, as well as the morphology and film microstructure through which ions traverse. Here, we present a straightforward technique that takes advantage of the electrochromic nature of conducting polymers in order to deduce links between conjugated polymer aggregates, molecular (dis)order, and ion penetration. The morphology of poly(3,4-ethylenedioxythiophene):poly(styrenesulfonate) (PEDOT:PSS) films and, subsequently, ionic transport are tuned by the addition of the co-solvent ethylene glycol in the dispersions. Time lapse UV-VIS spectra of these films, acquired during their redox-cycle, provide valuable insight into the pathways of ions inside the conducting polymer and the nature of aggregates therein. The structure–property relationships gained using such techniques promise to guide the design of polymeric active materials for devices that rely on mixed conduction.
- This article is part of the themed collections: 2022 Journal of Materials Chemistry Lectureship winner: Sahika Inal and Emerging Investigators 2016: Novel design strategies for new functional materials
 Please wait while we load your content...
Please wait while we load your content...

