
Palladium-catalyzed Si–C bond-forming silylation of aryl iodides with hydrosilanes: an enhanced enantioselective synthesis of silicon-stereogenic silanes by desymmetrization†
Abstract
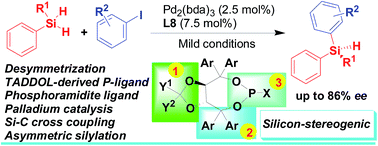
An enantioselective Pd-catalyzed silicon–carbon bond-forming silylation reaction of aryl iodides with hydrosilanes for the synthesis of silicon-stereogenic silanes has been developed, in which a systematic optimization of a TADDOL-derived monodentate phosphoramidite ligand set resulted in the identification of a new TADDOL-derived phosphoramidite ligand that accesses chiral silanes with moderate to good yield and enantioselectivity under mild conditions.
- This article is part of the themed collection: Editors’ collection: Catalytic Organic Transformations
 Please wait while we load your content...
Please wait while we load your content...

