
Introducing DDEC6 atomic population analysis: part 1. Charge partitioning theory and methodology†
Abstract
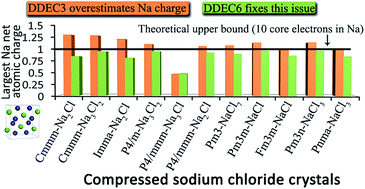
Net atomic charges (NACs) are widely used in all chemical sciences to concisely summarize key information about the partitioning of electrons among atoms in materials. The objective of this article is to develop an atomic population analysis method that is suitable to be used as a default method in quantum chemistry programs irrespective of the kind of basis sets employed. To address this challenge, we introduce a new atoms-in-materials method with the following nine properties: (1) exactly one electron distribution is assigned to each atom, (2) core electrons are assigned to the correct host atom, (3) NACs are formally independent of the basis set type because they are functionals of the total electron distribution, (4) the assigned atomic electron distributions give an efficiently converging polyatomic multipole expansion, (5) the assigned NACs usually follow Pauling scale electronegativity trends, (6) NACs for a particular element have good transferability among different conformations that are equivalently bonded, (7) the assigned NACs are chemically consistent with the assigned atomic spin moments, (8) the method has predictably rapid and robust convergence to a unique solution, and (9) the computational cost of charge partitioning scales linearly with increasing system size. We study numerous materials as examples: (a) a series of endohedral C60 complexes, (b) high-pressure compressed sodium chloride crystals with unusual stoichiometries, (c) metal–organic frameworks, (d) large and small molecules, (e) organometallic complexes, (f) various solids, and (g) solid surfaces. Due to non-nuclear attractors, Bader's quantum chemical topology could not assign NACs for some of these materials. We show for the first time that the Iterative Hirshfeld and DDEC3 methods do not always converge to a unique solution independent of the initial guess, and this sometimes causes those methods to assign dramatically different NACs on symmetry-equivalent atoms. By using a fixed number of charge partitioning steps with well-defined reference ion charges, the DDEC6 method avoids this problem by always converging to a unique solution. These characteristics make the DDEC6 method ideally suited for use as a default charge assignment method in quantum chemistry programs.
- This article is part of the themed collection: Computational chemistry
 Please wait while we load your content...
Please wait while we load your content...

