
Photocatalytic oxidation of small molecule hydrocarbons over Pt/TiO2 nanocatalysts†
Abstract
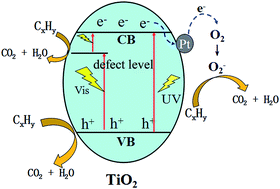
Highly active Pt/TiO2 catalysts were prepared by a simple photo-reduction method and used for catalytic oxidation of alkanes (C2H6 and C3H8) and alkenes (C2H4 and C3H6). It was found that significantly improved photo-activity can be reached even by loading a very small amount of Pt (0.2–0.5 wt%). Moreover, the Pt loading resulted in unexpected visible light activity for the oxidation of small molecule hydrocarbons. Further investigation using photoluminescence spectra indicate that the Pt loading helps reduce the charge carrier recombination within the TiO2 nanoparticles. Electron Paramagnetic Resonance (EPR) spectra reveal both oxygen molecules and lattice oxygen participate in the hydrocarbons' photooxidation. The transfer and reaction mechanisms of charge carriers during the photo-oxidation process are discussed in detail.
 Please wait while we load your content...
Please wait while we load your content...

