
Fermi level, work function and vacuum level
Abstract
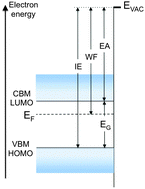
Electronic levels and energies of a solid, such as Fermi level, vacuum level, work function, ionization energy or electron affinity, are of paramount importance for the control of device behavior, charge carrier injection and transport. These levels and quantities, however, depend sensitively on the structure and surface morphology and chemical composition of the solid. A small amount of contaminants on a metal surface, or a shift in molecular orientation at the surface of an organic semiconductor, can change work function and vacuum level position by a large fraction of an electron-volt, and significantly impact the electronic structure of interfaces. The goal of this brief focus article is to provide definitions of key concepts and review simple mechanisms that affect these fundamental quantities.
- This article is part of the themed collections: Focus article collection and Materials Horizons 10th anniversary regional spotlight collection: The Americas
 Please wait while we load your content...
Please wait while we load your content...

