
Understanding conductivity in molecular switches: a real space approach in octaphyrins†
Abstract
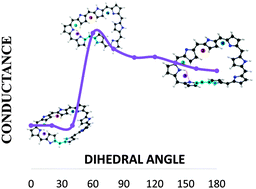
In recent years, expanded porphyrins have emerged as a promising class of π-conjugated molecules that display unique electronic, optical and conformational properties. Several expanded porphyrins can switch between planar and twisted conformations, which have different photophysical properties. Such a change of topology involves a Hückel–Möbius aromaticity switch in a single molecule and it can be induced by solvent, pH and metallation. These features make expanded porphyrins suitable for the development of a novel type of molecular switches for molecular electronic devices. Octaphyrins consisting of eight pyrrole rings, exhibit twisted-Hückel, Möbius and Hückel π-conjugation topologies depending on the oxidation and protonation state, with distinct electronic structures and aromaticity. Our working hypothesis is that a significant change in the conductance of expanded porphyrins will be observed after the topology switching. Despite the potential of Hückel–Möbius systems as conductance switches, the relationship between the conductance and the molecular topology is not yet understood. We have explored the performance of local descriptors of conductivity in simple molecules, as well as the relationship with conductance. Since these indexes provide a qualitative measure of delocalization and conductance in the probe molecules, we have carried out a local analysis of electrical conductance changes as a function of the π-conjugation in two examples. In one of them, the locality of the electronic changes ensures the ability of these indexes to describe the conductance as local. Moreover, it enables to identify which conformational switch would be more efficient from an electronic device perspective. However, we also show that it is not always possible to reduce conductance changes to one bond, and in those molecules where a deep rearrangement occurs far from the structural perturbation, local measures show a limited efficiency. This is a first step for the description of the connection between the molecular structure and conductance in molecular switches.
- This article is part of the themed collection: Electron delocalization and aromaticity: 150 years of the Kekulé benzene structure
 Please wait while we load your content...
Please wait while we load your content...

