
Sequential release of drugs from hollow manganese ferrite nanocarriers for breast cancer therapy†
Abstract
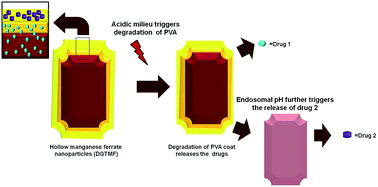
Single drug therapies for cancer are often suboptimal and may not provide long term clinical benefits. To overcome this obstacle for effective treatment the applications of two or more drugs are preferable. A limitation of multidrug use is the varying pharmacokinetics of different drugs. To overcome these impediments, we designed and synthesized multi-layered polyvinyl alcohol tethered hollow manganese ferrite nanocarriers capable of encapsulating two drugs with unique attributes of sensitivity towards tumor acidic milieu, mono-dispersive, compactness and high encapsulation efficiency. We encapsulated tamoxifen and diosgenin in the peripheral and subsequent inner layers of multilayered nanocarriers. In vitro and in vivo studies evaluated the nanocarrier uptake and retention ability of the tumor through magnetic saturation studies and elucidated the molecular mechanisms mediating drug(s)-induced apoptosis. The acidity of the tumor environment triggers extracellular dissociation of the peripheral coats resulting in release of tamoxifen blocking the estrogen receptor. The partially degraded nanocarriers localize intracellularly through endosomal escape and release diosgenin. Nanocarrier treatment reduced the cellular levels of Bcl2 and p53, while increasing the levels of Bim. This delivery system successfully embodies the sequential release of drugs and may provide a therapeutic strategy for sequentially affecting multiple targets in advanced cancers.
 Please wait while we load your content...
Please wait while we load your content...

