
Cu-Mediated 2,2,2-trifluoroethylation of terminal alkynes using 1,1-dichloro-2,2,2-trifluoroethane (HCFC-123)†
Abstract
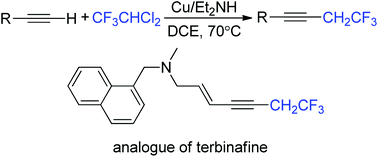
The title reaction provides a novel utilization of ozone-depleting/global-warming HCFCs as a new carbon–carbon cross-coupling model of various terminal alkynes by activating two inert C–Cl bonds successively. This protocol provided trifluoroethylated alkynes efficiently under mild reaction conditions and was compatible with a broad range of functional groups. An example of the synthesis of a terbinafine analogue is shown. Some mechanistic experiments including deuterated reagents and radical/SET inhibitors are described.
- This article is part of the themed collection: Celebrating 70 Years of Shanghai Institute of Organic Chemistry
 Please wait while we load your content...
Please wait while we load your content...

