
Transition metal-catalyzed C–H bond functionalizations by the use of diverse directing groups
Abstract
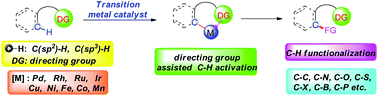
Transition metal-catalyzed direct functionalization of C–H bonds is one of the key emerging strategies that is currently attracting tremendous attention with the aim to provide alternative environmentally friendly and efficient ways for the construction of C–C and C–hetero bonds. In particular, the strategy involving regioselective C–H activation assisted by various functional groups shows high potential, and significant achievements have been made in both the development of novel reactions and the mechanistic study. In this review, we attempt to give an overview of the development of utilizing the functionalities as directing groups. The discussion is directed towards the use of different functional groups as directing groups for the construction of C–C and C–hetero bonds via C–H activation using various transition metal catalysts. The synthetic applications and mechanistic features of these transformations will be discussed, and the review is organized on the basis of the type of directing groups and the type of bond being formed or the catalyst.
- This article is part of the themed collection: Organic Chemistry Frontiers Outstanding Paper Awards 2014-2023
 Please wait while we load your content...
Please wait while we load your content...

