
Difluoromethyl 2-pyridyl sulfone: a versatile carbonyl gem-difluoroolefination reagent
Abstract
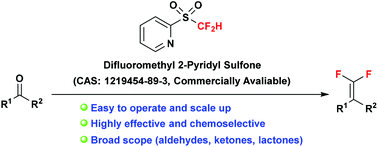
This tutorial account describes a robust carbonyl gem-difluoroolefination reagent, difluoromethyl 2-pyridyl sulfone (2-PySO2CF2H), developed by our group, recently. The guidelines for its laboratory preparation and application are presented in detail for potential users.
- This article is part of the themed collection: Celebrating 70 Years of Shanghai Institute of Organic Chemistry
 Please wait while we load your content...
Please wait while we load your content...

