
QM/MM MD simulations of La(iii)–phosphopeptide complexes
Abstract
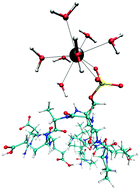
Several bioanalytical enrichment techniques are based on the interactions of phosphopeptides with Ln(III) ions. In order to gain an improved understanding of these complexes and the respective ion–peptide interactions, hybrid quantum mechanics/molecular mechanics (QM/MM) molecular dynamics (MD) simulations of La(III) coordinating to the phosphopeptide VPQLEIVPNSpAEER were conducted. Simulations of di- as well as monoanionic phosphate groups were carried out. The La(III) ion and its first hydration layer, including the sidechain of the phosphoserine residue were treated quantum mechanically at RI-MP2/triple zeta level, whereas the remaining part of the system was treated with classical potentials. The simulation of the dianionic phosphopeptide revealed a 9-fold coordinated La(III) ion, with the phosphopeptide binding bi- as well as monodentate. The mean residence times (τ) of the first shell water molecules were 82 ps and 37 ps for the bi- and monodentate complexes, respectively, which is much higher compared to free La(III) in aqueous solution (τ = 17 ps). The simulation of the monoanionic La(III)–phosphopeptide complex revealed a bidentate coordination throughout the 80 ps sampling period. An intramolecular hydrogen bond between the hydrogen of the phosphate group and the backbone was observed and a τ value of 14 ps was obtained, which is much lower as for the dianionic complex.
 Please wait while we load your content...
Please wait while we load your content...