
Rapid, targeted and culture-free viral infectivity assay in drop-based microfluidics†
Abstract
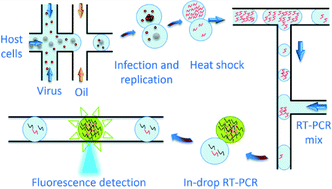
A key viral property is infectivity, and its accurate measurement is crucial for the understanding of viral evolution, disease and treatment. Currently viral infectivity is measured using plaque assays, which involve prolonged culturing of host cells, and whose measurement is unable to differentiate between specific strains and is prone to low number fluctuation. We developed a rapid, targeted and culture-free infectivity assay using high-throughput drop-based microfluidics. Single infectious viruses are incubated in a large number of picoliter drops with host cells for one viral replication cycle followed by in-drop gene-specific amplification to detect infection events. Using murine noroviruses (MNV) as a model system, we measure their infectivity and determine the efficacy of a neutralizing antibody for different variants of MNV. Our results are comparable to traditional plaque-based assays and plaque reduction neutralization tests. However, the fast, low-cost, highly accurate genomic-based assay promises to be a superior method for drug screening and isolation of resistant viral strains. Moreover our technique can be adapted to measuring the infectivity of other pathogens, such as bacteria and fungi.
- This article is part of the themed collection: Coronavirus articles - free to access collection
 Please wait while we load your content...
Please wait while we load your content...

